
Оглавление
- 1 Описание патологии
- 2 Сущность понятия
- 3 Наследуется ли фенилкетонурия
- 4 Симптомы заболевания
- 5 История
- 6 Разновидности фенилкетонурии
- 7 Патогенез
- 8 Признаки фенилкетонурии
- 9 Симптомы заболевания
- 10 Диагностика
- 11 Наследование
- 12 Классификация ФКУ по показателю фенилаланина в сыворотке
- 13 Лечение фенилкетонурии
- 14 Прогноз
- 15 Народная медицина для избавления от недуга
- 16 Питание при фенилкетонурии
- 17 Исторические данные
- 18 Заключение
Описание патологии
Развитие фенилкетонурии (ФКУ) связано с неспособностью организма человека с дефектным геном расщеплять поступающую вместе с пищей, богатой на белки, аминокислоту под названием фенилаланин.
В итоге кислота и элементы ее распада скапливаются в тканях и биожидкостях, превращаются в токсические соединения, под воздействием которых происходит глубокое и в большинстве случаев необратимое поражение органов ЦНС.
Отсутствие своевременного адекватного лечения становится причиной задержки развития интеллекта вплоть до олигофрении.
Манифестирует Фенилкетонурия обычно у больных малышей в первые 3-6 месяцев после рождения.
Выявление заболевания до появления первых клинических проявлений и соблюдение в дальнейшем эллиминационной диеты исключает развитие тяжелых, необратимых расстройств нервной системы у детей и взрослых, то есть опасности для физического, психоэмоционального и умственного здоровья нет.
Фенилкетонурия в среднем диагностируется у 1 новорожденного из 10 тысяч. В Турции количество больных большое — детей с ФКУ рождается 1 на 2, 5 тысячи, в Японии – 1 на 100 тысяч рожденных, в Африке (у представителей негроидной расы) болезнь встречается исключительно редко. У девочек патология диагностируется в 2 раза чаще по сравнению с мальчиками.
В медицинской литературе можно встретить и другие обозначения болезни — фенилпировиноградная олигофрения, болезнь Феллинга.
Сущность понятия
Фенилкетонурия является наследственным заболеванием, связано оно с серьезными нарушениями в белковом обмене. Это, в свою очередь, приводит к поражению нервной системы.
Неработоспособность всего одного фермента, фенилаланина, а в результате – такие серьезные проблемы со здоровьем, как фенилкетонурия. Что это за состояние, когда в организме происходит накопление большого количества токсических веществ? Все ядовитые соединения запасаются в биологических жидкостях, поэтому для медиков диагностировать болезнь обычно не составляет особого труда.

Если вовремя не принять меры, то можно наблюдать серьезные поражения нервной системы, а это уже приводит к нарушениям в функционировании всего организма.
Таким образом, без соответствующего лечения о нормальной жизни пациента не может быть и речи.
Наследуется ли фенилкетонурия
Заболевание передается по наследству, однако только в том случае, если оба супруга (биологические родители) являются носителями дефективного гена. Но патология у детей таких пар развивается не всегда:
- В 25 % на свет появляются больные малыши;
- В 50 % случаев дети являются носителями дефектного гена фенилкетонурии;
- В 25 % рождаются абсолютно здоровые по всем показателям малыши.
Наличие гена ФКУ определяется в медико-генетических центрах. Если у пары уже рожден ребенок, больной фенилкетонурией, то обследование для них обязательно перед планированием повторной беременности.
Среди населения носителей патологического гена насчитывается 2-3 %, но заболевание встречается относительно редко в связи с тем, что для его развития необходимо, чтобы и у папы, и у мамы был ген, вызывающий данную болезнь.
Симптомы заболевания
В основе заболевания лежит генетический дефект – мутация гена 12-й хромосомы (98% всех случаев фенилкетонурии). Это так называемая классическая фенилкетонурия. Ген кодирует количество фермента фенилаланин-4-гидроксилазы. Фермент отвечает за превращение аминокислоты фенилаланина в организме человека в тирозин в клетках печени. Фенилаланин – это аминокислота, которая содержится в белковых продуктах (мясо, рыба, молоко, яйца и другие).
При мутации гена количество фермента снижается, что приводит к накоплению фенилаланина и продуктов промежуточного метаболизма в тканях организма. Организм пытается избавиться от фенилаланина и продуктов его распада, выводя их с мочой.
Подобные нарушения обмена веществ приводят к нарушению строения нервных проводников, снижению образования нейромедиаторов. Все это, наряду с прямым токсическим действием избытка фенилаланина, приводит к развитию умственных нарушений, являющихся основным проявлением заболевания.
Фенилкетонурия наследуется по аутосомно-рецессивному типу, то есть не зависит от пола, и возникает при совпадении двух патологических генов от отца и матери.
Остальные 2% случаев фенилкетонурии связаны с другими генетическими дефектами и зависят от концентрации прочих ферментов (дигидроптеридинредуктазы и др.). Они имеют те же клинические проявления, но не поддаются лечению диетой. Такие варианты относят к атипичному течению заболевания. Среди них принято выделять фенилкетонурию II и III. Генетический дефект при фенилкетонурии II располагается в 4-й хромосоме, при III – в 11-й хромосоме.
Частота данного заболевания в разных странах отличается в разы. Так, в России рождается один больной малыш на 10 тысяч новорождённых. Этот показатель в некоторых регионах Великобритании в два раза выше – 1:5000. На Африканском же континенте дети почти не болеют фенилкетонурией. Также известно, что количество девочек среди всех больных практически вдвое превышает количество мальчиков.
Среди всех наследственных болезней фенилкетонурия является почти единственной, которую полностью удаётся нейтрализовать. На сегодняшний день такого ребёнка можно вырастить абсолютно здоровым.
В 98% всех случаев фенилкетонурии в её основе лежит генетический дефект, а именно мутация гена XII хромосомы, который кодирует количество фермента фенилаланин-4-гидроксилазы, отвечающего за превращение аминокислоты фенилаланина в клетках печени в тирозин. В результате снижается количество фермента, что приводит к накоплению аминокислоты и продуктов её промежуточного обмена в тканях.
Вследствие побочных путей обмена фенилаланин превращается в вещества, которых в организме не должно быть в норме: фенилмолочную и фенилпировиноградную кислоты, ортофенилацетат и фенилэтиламин. Они накапливаются в крови больного и оказывают комплексное действие:
- вызывают дефицит нейромедиаторов, передающих между клетками нервной системы нервный импульс;
- нарушают процессы жирового обмена в головном мозге;
- отравляют мозг, оказывая токсическое действие.

Это вызывает необратимое снижение интеллекта. У такого ребёнка достаточно быстро развивается олигофрения.
Остальные 2% случаев заболевания связаны с другими генетическими нарушениями и зависят от концентрации иных ферментов (дигидроптеридинредуктазы и др.). Для них характерны те же самые клинические проявления, однако они не поддаются лечению диетой.
В основе заболевания лежит генетический дефект – мутация гена 12-й хромосомы (98% всех случаев фенилкетонурии). Это так называемая классическая фенилкетонурия.
Ген кодирует количество фермента фенилаланин-4-гидроксилазы. Фермент отвечает за превращение аминокислоты фенилаланина в организме человека в тирозин в клетках печени.
Фенилаланин – это аминокислота, которая содержится в белковых продуктах (мясо, рыба, молоко, яйца и другие).
Они имеют те же клинические проявления, но не поддаются лечению диетой. Такие варианты относят к атипичному течению заболевания. Среди них принято выделять фенилкетонурию II и III.
Генетический дефект при фенилкетонурии II располагается в 4-й хромосоме, при III – в 11-й хромосоме.
Симптомы
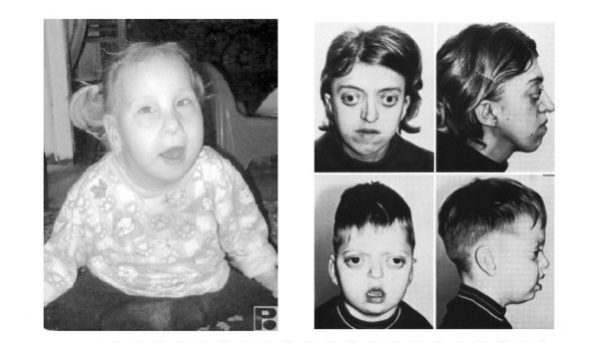
У детей с фенилкетонурией светлые волосы, бледная кожа и голубые глаза.
Ребенок с фенилкетонурией рождается внешне здоровым, то есть ничем не отличается от других детей. С поступлением пищи в организм начинается попадание белка, а значит и фенилаланина. Последний постепенно накапливается, и обычно к 2 месяцам жизни появляются первые симптомы: вялость или беспокойство, отсутствие интереса к окружающему миру, срыгивания, изменения мышечного тонуса.
К 4-6 месяцам становится очевидной задержка психического развития. Ребенок не следит за игрушкой, не реагирует на звук, не узнает родителей.
Чем дольше продолжается поступление фенилаланина в организм с едой, тем выраженнее нарушения в психической и мыслительной сферах. Развитие речи резко задерживается. Иногда словарный запас может ограничиваться несколькими словами.
Если диагноз не будет выставлен и не будет начато лечение, то к 3-4 годам умственные нарушения достигнут степени идиотии (самая тяжелая степень олигофрении).
Особенностью клинического течения фенилкетонурии является необратимость возникших психических и интеллектуальных изменений. То есть при позднем выявлении помочь таким деткам уже нельзя – на всю жизнь они остаются умственно отсталыми.
Физическое развитие также отстает: дети позже начинают держать голову, переворачиваться, сидеть. Когда такие дети начинают ходить, то при этом они широко расставляют ножки, сгибая их одновременно в коленных и тазобедренных суставах.
Походка покачивающаяся, мелкими шажками. В положении сидя дети принимают «позу портного» — сгибают и руки, и ноги, поджимая последние под себя. Обычно объем головы меньше, чем в норме. Может быть выраженная микроцефалия: маленькая голова.

Из других неврологических симптомов возможны нарушения мышечного тонуса, судорожные припадки. Эпилептические приступы обычно появляются в возрасте 1,5 лет и приводят к еще большему прогрессированию нарушений интеллекта.
У части больных фенилкетонурией появляются непроизвольные движения в конечностях, дрожание (гиперкинезы). В движениях нет плавности и согласованности, нарушается равновесие.
Кроме ряда психических и интеллектуальных изменений, фенилкетонурию характеризуют следующие симптомы:
- специфический «мышиный» запах (или запах плесени) от ребенка: этот симптом характерен только для фенилкетонурии. Запах появляется в результате выделения продуктов метаболизма фенилаланина (фенилпировиноградной, фенилмолочной, фенилуксусной кислот) через кожу и с мочой;
- кожные проявления: дерматиты, экзема, просто шелушение (возникают по той же причине, что и «мышиный» запах);
- позднее прорезывание зубов: у таких детей первые зубы могут появиться после 18 месяцев, эмаль недоразвита;
- нарушение пигментации: у таких детей обычно голубые глаза, очень светлая кожа и волосы в результате снижения количества меланина (его содержание зависит от метаболизма фенилаланина). Из-за этого у таких детей наблюдается повышенная чувствительность к солнечному свету;
- вегетативные симптомы: пониженное артериальное давление, повышенная потливость, запоры, акроцианоз (синюшность кистей и стоп);
- нередко фенилкетонурия сопровождается врожденными пороками сердца.
Атипичные случаи фенилкетонурии, связанные с нарушением деятельности других ферментов, участвующих в метаболизме фенилаланина, кроме умственных изменений характеризуются развитием мышечной слабости во всех конечностях с одновременным повышением мышечного тонуса, спастическим тетрапарезом. Также при этих формах развивается слюнотечение, приступы повышения температуры.
У взрослых людей, страдающих фенилкетонурией, возможно появление судорожных припадков, нарушений координации, дрожания в конечностях, ухудшения памяти и внимания, возникновение депрессии. Обычно подобные симптомы возникают при несоблюдении элиминационной диеты.
Диагностика
Скрининговые тесты на содержание фенилаланина всем новорожденным проводят в роддоме.
В связи с тем, что фенилкетонурия сопровождается развитием необратимых умственных нарушений, во многих странах мира, в том числе и в России, принято использовать скриниг-методы диагностики. Что это означает? Всем без исключения новорожденным детям в роддоме проводят экспресс-тесты на содержание фенилаланина.
Для этого берут капиллярную кровь (из пятки) на 4-5-й день жизни ребенка (у недоношенных на 7-й), наносят на специальный бумажный бланк и отправляют в лабораторию, где по определенным изменениям врач-лаборант делает выводы о содержании фенилаланина в крови. Отрицательный тест говорит об отсутствии фенилкетонурии.

Если тест оказывается положительным, то тогда проводят дополнительные исследования для определения содержания фенилаланина в крови и моче (хроматографию, флюориметрию). Концентрацию фенилаланина в крови и моче регулярно проверяют при проведении лечения, чтобы контролировать эффективность диеты и корригировать ее при необходимости.
Возможно проведение генетического исследования для подтверждения мутации в гене, отвечающем за фенилаланин-4-гидроксилазу.
Подобное исследование возможно в качестве пренатальной диагностики, то есть на этапе беременности (берут околоплодные воды путем пункции).
Это инвазивное исследование делают по строгим показаниям (например, наличие больного фенилкетонурией ребенка в семье). Выявление генетического дефекта у плода позволяет прервать беременность.
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин. Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека.
Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются. Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ.
История
Фенилкетонурия как болезнь, протекающая с задержкой интеллектуального и психического развития, открыта в 1934 году норвежским ученым-исследователем Иваром Асбьером Феллингом, отсюда и другое обозначение патологии — болезнь Феллинга.
Первых успехов в коррекции здоровья малышей с ФКУ добились медики во главе с Хорстом Биккелем в середине прошлого века. Разработка терапии и ее внедрение проводились на базе Бирмингемского детского госпиталя (Англия).

Однако больших положительных результатов ученым удалось добиться, когда начала широко проводится ранняя диагностика новорожденных на выявление фенилкетонурии, это примерно 1958-1961 год прошлого века.
Расширение возможностей диагностики со временем позволило установить, что в развитии заболевания принимает участие только ген фенилаланингидроксилазы (РАН).
За последние десятилетия описаны атипичные варианты течения болезни, разработаны и широко применяются новейшие способы ее коррекции. В перспективе – использование генотерапии, что вполне вероятно позволит полностью победить болезнь.
Разновидности фенилкетонурии
Если рассматривать формы заболевания, то они могут быть такими:
- Классическая. В этом случае мы наблюдаем, что фенилкетонурия – рецессивный признак. Встречается такая форма у одного ребенка на десять тысяч здоровых детей. Если не принять меры, то едва ли больной человек доживет до тридцати лет.
- Вариативная форма. Не передается по наследству, а вызывает ее мутация в генах. Течение ее более тяжелое, а ранняя смертность прогнозируется с вероятностью практически 100%.
Кроме форм, врачи различают еще и типы фенилкетонурии:
- Первый тип характеризуется тем, что наблюдается недостаток фермента фенилаланин-4-гидроксилазы, который и отвечает за превращение фенилаланина. В 98% случаев диагностируется именно он.
- Второй. Его отличает малое содержание фермента дигидроптеридинредуктазы. У таких больных наблюдаются судороги, а также умственная отсталость. Несмотря на свою редкую встречаемость, смертность от этого типа может наступать в 2-3-летнем возрасте.
- Третий тип характеризуется тем, что возникает дефицит тетрагидробиоптерина. В результате происходит уменьшение объемов мозга, что приводит к умственной отсталости.
Выделяют две формы заболевания:
- Классическая. В данном случае фенилкетонурия является рецессивным признаком. Она встречается у одного ребёнка примерно на десять тысяч здоровых детей. Если не принимать никаких мер, то больной человек едва ли доживёт и до тридцати лет.
- Вариативная. Она не передаётся по наследству, а вызывает её генетическая мутация. Течение заболевания в этом случае более тяжёлое, а ранняя смертность прогнозируется почти со стопроцентной вероятностью.

В зависимости от дефекта гена, который блокирует определённый фермент, принято выделять три типа патологии:
- Фенилкетонурия I типа (классическая). Вызывается генетической мутацией, которая нарушает выработку в печени фермента фенилаланингидроксилазы и превращение фенилаланина в тирозин. В 98% всех случаев заболевания диагностируется именно она.
- Фенилкетонурия II типа характеризуется генетическим дефектом, вызывающим дефицит фермента дигидробиоптеринредуктазы. В результате нарушается активность органического соединения, которое необходимо для преобразования фенилаланина. Кроме того, наблюдается также пониженное содержание витамина В9 в сыворотке крови и в спинномозговой жидкости, а он необходим для утилизации аминокислот. У больных отмечаются судороги и умственная отсталость. Смертность от данного типа болезни может наступать в двух- трёхлетнем возрасте.
- Фенилкетонурия III типа провоцируется дефицитом катализатора, нужного для синтеза тетрагидробиоптерина, который необходим для превращения фенилаланина в тирозин. Вследствие этого происходит уменьшение объёмов головного мозга, что приводит к умственной отсталости.
Существует также так называемая примаптеринурия – это атипичное течение болезни, которое возникает при лёгкой форме гиперфенилаланинемии. В настоящее время ферментный дефект этого вида фенилкетонурии не выяснен. Однако данная форма патологии характеризуется высоким количеством в моче примаптерина и его производных, при этом содержание в спинномозговой жидкости нейромедиаторных метаболитов не отклоняется от нормы.
Ещё выделяют материнскую фенилкетонурию, которая наблюдается у потомства женщин с данной патологией, не соблюдающих специальную диету. Механизм развития этой формы заболевания не изучен до конца, но известно, что без регулярного контроля уровня фенилаланина у новорождённых детей выявляется ряд патологических изменений:
- недостаточный вес головного мозга;
- недоразвитие белого вещества;
- вентрикуломегалия (увеличение размеров желудочков мозга).
Фенилкетонурия у детей этого типа провоцирует хроническую интоксикацию у плода и приводит к умственной отсталости.
Патогенез
При фенилкетонурии в печени больного не продуцируется особый фермент под названием фенилаланин-4-гидроксилаза.
Основная его функция – преобразование поступающего в органы ЖКТ с пищей фенилаланина в аминокислоту тирозин. Это аминокислота входит в состав большинства ферментов, гормонов, способствует выработке меланина (пигмент), принимает участие в образовании белков и в функционировании большинства внутренних органов.
Метаболический блок при фенилкетонурии неуклонно приводит к тому, что начинают работать побочные (атипичные) пути обмена фенилаланина, в результате он трансформируется в те вещества, которых в теле здорового человека быть не должно.
Это кислоты – фенилмолочная и фенилпировиноградная, ортофенилацетат, фенилэтиламин. По сути, они являются токсическими соединениями, и их накапливание в кровеносной системе приводят:
- К нарушению нормального обмена липидов (жировых клеток) в разных отделах головного мозга;
- К нехватке нейромедиаторов, ответственных за бесперебойную передачу нервных импульсов во всей нервной системе.
В результате это приводит к прогрессирующему снижению интеллекта и к умственной отсталости выраженной степени – олигофрении, имбецильности, идиотии.
Отсутствие терапии становится причиной того, что через каждые 10 недель коэффициент развития интеллекта больного малыша падает на 5 пунктов.
Признаки фенилкетонурии
Приблизительно к четырём-шести месяцам становится заметной задержка психического развития. Малыш не узнаёт родителей, не реагирует на звук, не следит за игрушкой. Чем дольше длится поступление в организм фенилаланина с едой, тем выраженнее нарушения. Резко задерживается развитие речи. Словарный запас иногда может ограничиваться лишь несколькими словами.
Особенностью фенилкетонурии является необратимость интеллектуальных и психических изменений. Таким образом, помочь таким деткам при позднем выявлении уже нельзя – они на всю жизнь остаются умственно отсталыми.
Также заметно отстаёт и физическое развитие: такие дети позже начинают переворачиваться, держать голову, сидеть. При становлении ходьбы они широко расставляют ножки, одновременно сгибая их в тазобедренных и коленных суставах. Походка у них покачивающаяся, мелкими шажками. Дети принимают так называемую «позу портного» в положении сидя — сгибают все конечности, поджимая ноги под себя. Объём головы обычно меньше нормы. Может наблюдаться выраженная микроцефалия.
Из иных неврологических симптомов возможны изменения мышечного тонуса, а также судороги. Эпилептические приступы, как правило, появляются примерно в возрасте полутора лет и приводят к прогрессированию умственных нарушений. У некоторых больных отмечаются непроизвольные движения в конечностях. В их движениях нет согласованности и плавности, нарушается равновесие.
Помимо ряда интеллектуальных и психических изменений для фенилкетонурии характерны следующие проявления:
- специфический «мышиный» запах от ребёнка (он характерен только для фенилкетонурии и появляется из-за выделения через кожу и с мочой продуктов метаболизма фенилаланина: фенилуксусной, фенилмолочной, фенилпировиноградной кислот);
- кожные проявления: шелушение, экзема, дерматиты (возникают по той же причине);
- позднее прорезывание зубов (первые зубы у таких детей могут появиться после полутора лет, эмаль при этом недоразвита);
- нарушение пигментации: обычно у таких детей голубые глаза, светлая кожа и волосы из-за снижения количества меланина;
- вегетативные симптомы: повышенная потливость, пониженное артериальное давление, запоры;
- нередко выявляются врождённые пороки сердца.
У взрослых возможно появление дрожания в конечностях, нарушений координации, судорожных припадков, ухудшения внимания и памяти, возникновение депрессии. Подобные симптомы обычно возникают при несоблюдении специализированной диеты.
У детей с фенилкетонурией бледная кожа, очень светлые волосы и голубые глаза.
Постепенно при отсутствии надлежащего лечения у больного можно будет наблюдать такие признаки:
- Судорожный синдром. Он начинает проявляться в раннем детстве и сохраняется у взрослых.
- Недостаток пигмента в коже и волосах. Поэтому такие пациенты, как правило, светловолосые и имеют белую кожу.
- Воспалительные процессы, которые по незнанию можно принять за аллергическую реакцию.
Первые признаки умственного отставания можно заметить у ребенка уже в возрасте полугода. Он перестает запоминать новую информацию, и кажется, что он совершенно не способен к обучению. Насторожиться родители должны и тогда, когда малыш забывает то, чему уже давно научился, например, как держать ложку, сидеть, играть с погремушкой.
Вот признаки, которые имеет фенилкетонурия, симптомы болезни необходимо рассматривать только в комплексе, потому что по отдельности они вполне могут встречаться и у здоровых детей.
Симптомы заболевания
К 4-6 месяцам становится очевидной задержка психического развития. Ребенок не следит за игрушкой, не реагирует на звук, не узнает родителей. Чем дольше продолжается поступление фенилаланина в организм с едой, тем выраженнее нарушения в психической и мыслительной сферах. Развитие речи резко задерживается.

Физическое развитие также отстает: дети позже начинают держать голову, переворачиваться, сидеть. Когда такие дети начинают ходить, то при этом они широко расставляют ножки, сгибая их одновременно в коленных и тазобедренных суставах. Походка покачивающаяся, мелкими шажками. В положении сидя дети принимают «позу портного» — сгибают и руки, и ноги, поджимая последние под себя. Обычно объем головы меньше, чем в норме. Может быть выраженная микроцефалия: маленькая голова.
Дети, больные фенилкетонурией, внешне на первых неделях жизни ничем не отличаются от здоровых. Поступление пищи (материнского молока или смесей) приводит к накапливанию у них в организме фенилаланина и побочных (токсических) продуктов его обмена. И обычно к 2-3 месяцем появляются первые признаки патологии, это:
- Вялость;
- Возбудимость или наоборот заторможенность;
- Частые срыгивания, иногда переходящие в упорную рвоту, что может быть расценено за симптом пилоростеноза;
- Судороги после 3 месяцев.
Из-за срыгивания и рвоты малыши не добирают в весе. Если на этом этапе начать лечение, то дальнейших патологических изменений в организме происходить не будет.
При отсутствии адекватной терапии ближе к 4-5 месяцам появляются первые признаки задержки психического развития:
- Отсутствие интереса к окружающей действительности;
- Младенец не распознает родителей, не обращает внимания на яркие игрушки, предметы, мало реагирует на звуки;
- Задерживается речевое развитие.
При фенилкетонурии страдает и уровень физического развития. Такие дети гораздо позже по сравнению со здоровыми сверстниками начинают активно переворачиваться, удерживать головку, сидеть.

Обратить внимание можно и на своеобразную походку – ножки широко расставлены в стороны, одновременно согнуты коленные и тазобедренные суставы. Это приводит к появлению шатающейся походки.
В позе сидя у детей скрещены и подогнуты под себя ноги и согнуты в локтевых суставах руки (поза портного). В движениях нет мягкости и плавности, нередко отмечается дрожание конечностей, гиперкинезы.
Возможно нарушение тонуса мышц, у части малышей к полутора-двум годам возникают эпилептические припадки.
При фенилкетонурии умственные и психические изменения, уже возникшие на момент начала терапии, скорректировать невозможно. Поэтому при позднем лечении у детей будет в то или иной мере наблюдаться задержка интеллектуального развития.
В тяжелых случаях к 4 годам умственные нарушения приобретают вид идиотии – самой запущенной стадии олигофрении.
Помимо интеллектуальных и психических отклонений заболевание проявляется и другой симптоматикой:
- Специфическим, неприятным (некоторые называют его мышиный или плесневый) запахом от больного малыша. Характерен только для фенилкетонурии. Возникает вследствие выделения продуктов расщепления фенилаланина с мочой и через поры кожи;
- Изменения на кожи – появление шелушащихся пятен, развитие экземы и дерматитов. Возникают они также как следствие выхода токсических соединений через кожу;
- Поздно (к 15-18 месяцев) прорезываются зубы, наблюдается гипоплазия эмали;
- Гипопигментация. Снижение меланина становится причиной постепенного посветления кожи, волос и глаз. Обычно дети с ФКУ светлокожие с голубыми зрачками и белыми волосами, поэтому у них повышена чувствительность к солнечным лучам;
- Гипотонией, чрезмерной потливостью, периодическими запорами, синюшностью кожи кистей рук и стоп;
- Врожденными пороками сердечной мышцы.
При II типе ФКУ наблюдается сухожильная гиперрефлексия, судороги, спастический тетрапарез, тяжелая степень отклонений в интеллектуальном развитии.
Заболевание обычно быстро прогрессирует, и вследствие необратимых изменений гибель малыша происходит на третьем-четвертом году жизни.

Для ФКУ III характерно – микроцефалия, тетрапарез спастический, олигофрения.
Злокачественная форма болезни характеризуется:
- Быстрым прогрессированием неврологических нарушений, в том числе и умственной отсталости (при этом уровень ФА вследствие диетотерапии может быть в пределах нормы);
- Нарушением глотания;
- Выраженным беспокойством малыша;
- Тяжело купирующимися судорожными приступами.
При злокачественной ФКУ нервная система быстро повреждается и если вовремя не будет проведено соответствующее лечение, то смерть малыша наступит в течение нескольких месяцев.
Отказ от лечебного питания взрослых пациентов приводит к колебанию настроения, к внутреннему беспокойству, страху и депрессии, что создает социальные проблемы.
На высокий уровень фенилаланина после прекращения лечения указывает:
- Бессонница;
- Импульсивность;
- Деконцентрация;
- Потеря способности к рациональной оценке происходящих событий.
Диетотерапия необходима даже, если интеллект уже снижен. Соблюдение правильного питания позволяет снизить агрессию у больных, уменьшает чувство тревожности и улучшает контактирование с окружающими.
Нелеченая фенилкетонурия приводит к глубокой степени умственной отсталости, в большинстве случаев это имбецильность или идиопатия.
Не исключается развитие эхопраксии – повторение движений за окружающими людьми, эхолалии – повторений слов. Характерна также вялость с периодическими явлениями раздражительности, агрессии и злобы.
- Частая интенсивная рвота.
- Необоснованное беспокойство.
- Вялость, слабые двигательные реакции.
- Рассеянный блуждающий взгляд.
- Отсутствие эмоций.
К полугодовалому возрасту малыш не узнаёт маму, у него задерживается психомоторика, нарушается мышечный тонус, отсутствует оживлённая реакция на окружающий мир. Со временем симптомы только нарастают. У детей старше 3 лет уже замечаются:
- Психотические расстройства.
- Расторможенность.
- Нарушения поведения.
- Быстрая утомляемость.
- Чрезмерная возбудимость.
- Физическое и умственное отставание вплоть до идиотии.

У детей запаздывают физиологические процессы: прорезывание зубов, способность переворачиваться, сидеть. У них:
- Специфическая стойка в виде широко расставленных ног, опущенных плеч и головы.
- Походка покачивающаяся, шаги маленькие.
- Возможны непроизвольные движения, тремор с развитием эпилепсии.
- Гипергидроз, кисти рук и стопы синюшного цвета.
- Моча и пот с «мышиным» запахом из-за содержащейся в них фенилуксусной кислоты.
Без лечения не избежать экзематозных изменений на коже, фолликулярного кератоза, гнойных инфекций, возникновения судорог разной степени интенсивности. Постепенно присоединяются: гиперактивность, аномалии в работе зрительного анализатора, паркинсонизм, выполнение бесцельных движений, глубокая задержка моторики — каждый третий малыш не ходит, 60% не говорят.
- частая рвота без видимых причин;
- плаксивость;
- вялость;
- могут появляться высыпания по всему телу;
- моча имеет «мышиный» запах;
- ребенок отстает в физическом и умственном развитии от своих сверстников.
Достаточно взять анализ крови и мочи, чтобы поставить правильный диагноз.
Диагностика
Для ребёнка важно уже с первых дней жизни приобретать различные навыки, без которых невозможно нормальное развитие. Раннее выявление ФКУ I обеспечивает полную реабилитацию малыша, его адаптацию в окружающем мире. Благодаря методике скрининга фенилкетонурии здоровье новорождённого находится под контролем вне зависимости от региона России, где тот появился на свет. В программе задействованы все детские медучреждения, роддомы, медико-генетические центры.
Суть скрининг-теста состоит в заборе на 4—5-й день жизни младенца нескольких капель крови для анализа по выявлению ФА и тирозина. Биологическая жидкость исследуется при помощи радиоиммунного метода. Результаты в виде соответствующего штампа заносятся в обменную карту. Ошибка может возникнуть при недоношенности ребёнка. Для уточнения диагноза или его опровержения проводится повторный анализ. Менее точные результаты получаются при тестировании мочи.
При положительном итоге врач приглашает родителей для беседы и незамедленного начала лечебных мероприятий. Может понадобиться помощь медицинского генетика и эндокринолога для назначения пробы Феллинга, теста Гатри, хроматографии, флуориметрии, МРТ и ЭЭГ-исследования. При поиске дефектного гена берутся образцы крови или слюны родителей.
Для ФКУ II характерно прогрессирование болезни вне зависимости от диетотерапии. Патология сопровождается выраженной умственной отсталостью, судорогами, гиперрефлексией. К 2—3 годам возможен летальный исход. При ФКУ III наблюдается триада симптомов:
- Спастический тетрапарез.
- Микроцефалия.
- Глубокая умственная отсталость.

У каждой формы свой патогенез, клиническая картина и методы лечения. Для атипичных вариантов ФКУ (до 1% от всех случаев) оно сложное из-за устойчивости к диетотерапии. Поэтому сначала определяют тип заболевания, детально исследовав биохимию патологии.
Возможно проведение генетического исследования для подтверждения мутации в гене, отвечающем за фенилаланин-4-гидроксилазу. Подобное исследование возможно в качестве пренатальной диагностики, то есть на этапе беременности (берут околоплодные воды путем пункции). Это инвазивное исследование делают по строгим показаниям (например, наличие больного фенилкетонурией ребенка в семье). Выявление генетического дефекта у плода позволяет прервать беременность.
В случае если имеется подозрение, что кто-то из родителей является носителем гена фенилкетонурии, то это можно определить при помощи генетической экспертизы в медико-генетических центрах.
Дефект ещё на этапе беременности может быть обнаружен в результате инвазивных методов пренатальной диагностики (биопсия хориона, амниоцентез, кордоцентез).
После рождения всем новорождённым проводится так называемый неонатальный скрининг. Данная процедура является эффективным методом выявления самых распространённых наследственных заболеваний: адреногенитальный синдром, врождённый гипотиреоз, галактоземия, муковисцидоз и фенилкетонурия.
У каждого ребёнка в роддоме берут несколько капель крови из пяточки. Взятие образца проводят через три часа после кормления на четвёртый день жизни у доношенных детей и на седьмой день — у недоношенных. Кровь наносится на специальный тест-бланк, который затем отправляется в лабораторию, где и проводится генетическое исследование.

В случае если в данном анализе выявляют дефектный ген, то родителей с малышом приглашают для обследования в медико-генетический центр. С целью опровержения или подтверждения диагноза используются дополнительные исследования:
- в сыворотке крови;
- в сухом пятне крови;
- копрограмма;
- потовый тест;
- ДНК-диагностика.
Для подтверждения фенилкетонурии определяется концентрация в крови фенилаланина и тирозина, активность печёночных ферментов (фенилаланингидроксилазы), проводится биохимическое исследование мочи с целью обнаружения кетоновых кислот, метаболитов катехоламинов и др.
Дополнительно используются электроэнцефалография (ЭЭГ) и магнитно-резонансная томография (МРТ) головного мозга, а также осмотр ребёнка детским неврологом.
Уважаемые родители!
Фенилаланин (ФА) — это эгзогенная незаменимая аминокислота, необходимая для нормального роста и развития, которая поступает в организм с пищей. У пациентов с ФКУ доза фенилаланина ограничивается до количества, которое зависит от индивидуальной толерантности к ФА.
Низкобелковая диета позволяет удерживать концентрацию ФА в сыворотке крови больного уровне на безопасном для ЦНС уровне. Этот уровень определен для каждой возрастной группы. Для грудного возраста ФА должен быть на уровне 2-4мг%, проверка уровня концентрации ФА в сыворотке крови пациента проводится 1 раз/в неделю до 6 месяца жизни.
Чтобы содержание фенилаланина находилось на определённом „безопасном” уровне, диета должна состоять из лечебных препаратов с низким содержанием фенилаланина или без него (которые удовлетворяют потребность в белке на 70-80%), и такого количества натуральных продуктов, чтобы удовлетворить потребности организма в белке, минеральных компонентах, витаминах и фенилаланине, учитывая основные возрастные потребности ребёнка.
- Уменьшение дозы фенилаланина согласно индивидуальной толерантности фенилаланина, что означает уменьшение дозы натурального белка в суточном рационе
- Обеспечение соответствующей для нормального развития дозы белка (дополнительный белок без фенилаланина) из продуктов лечебного питания ФКУ
- Обеспечение соответствующей дозы энергии с использованием специальных низкобелковых продуктов
- Обеспечение соответствующей дозы витамин, макро- и микроэлементов – главным образом из препаратов ФКУ и других источников.
|
|
|
У пациентов с ФКУ количество потребляемого белка из натуральных продуктов не может превысить установленной нормы. В связи с этим у маленьких детей и у старших преобладающая часть потребности в белке, т.е. около 80%, должно быть погашено смесями, не содержащими фенилаланин, обогащёнными минеральными ингредиентами.

Диета грудного ребёнка с ФКУ базируется на смесях (препаратах) без фенилаланина, которые являются главными источниками белка, витаминов, микро- и макроэлементов. Грудное молоко и молочная смесь для младенцев дополняют эту диету необходимым для роста фенилаланином. Количество препарата ФКУ, грудного молока или молочных смесей надо систематически изменять в зависимости от индивидуальной толерантности фенилаланина, а также и от потребностей растущего организма.
только смесь аминокислот («белковый эквивалент») в сочетании с витаминами и минералами. Необходимое количество белка свободного от фенилаланина поступает в организм из низкоэнергетических напитков: фруктовые соки, фруктово-овощные соки. Такая смена режима и рациона питания может повлиять на обмен аминокислот.
В связи с тем, что смеси свободные от фенилаланина содержат мало калорий, а питание должно быть сбалансированным, т.е. отвечать определённым соотношениям содержания жиров и углеводов (основных источников энергии) таким образом, чтобы полностью удовлетворить суточную потребность организма в энергии, целесообразным становится обогащение суточного рациона иными высокоэнергетическими пищевыми прдуктами. Это стало возможным благодаря наличию на рынке большого количества специализированных низкобелковых и частично свободных от фенилаланина продуктов.
Дневная доза смеси ФКУ зависит от возраста ребёнка, массы тела, общего состояния здоровья и индивидуальной суточной толерантности фенилаланина. Очень важно, чтобы рекомендуемое суточное количество смеси не давать в один приём, напр.
утром. Такой способ подачи смеси может привести к колебаниям аминокислотного равновесия или к симптомам нетолерантности препарата. Суточную дозу смеси необходимо поделить на 3-4 приёма в течении дня. Препарат следует принимать во время еды.
Суточное потребление фенилаланина из пищевых продуктов должно быть ограничено до такого количества, чтобы контролируемый уровень концентрации фенилаланина в сыворотке крови не превышал „безопасного для ЦНС” уровня, т.е.
Наследование
Так как фенилкетонурия наследуется как рецессивный признак, то для проявления его у ребенка необходимо, чтобы у обоих родителей имелся дефектный ген. Именно поэтому родственные браки во многих странах запрещены.
Если рассматривать случай рождения детей в обычной семье, то у носителей такой мутации они могут быть:
- 25% вероятности, что ребенок родится больным.
- В 50% случаев малыш здоров, но является носителем дефектного гена.
- Четвертая часть потомства будет абсолютно нормальной.
Эта схема не дает полной картины рождаемости больных детей. Она только отражает вероятность, поэтому у каждой семейной пары процент дефектных генов может быть свой, и предсказать исход, к сожалению, невозможно. Сейчас существуют консультации, в которых специалисты-генетики помогают парам спрогнозировать рождение у них больного ребенка, рассказывая при этом, как фенилкетонурия наследуется.
Классификация ФКУ по показателю фенилаланина в сыворотке
В некоторых странах фенилкетонурия классифицируется на типы в зависимости от уровня фенилаланина в сыворотке крови:
- Классическая ФКУ – показатель фенилаланина достигает 20 мг% (1200 мкмоль/л) и выше;
- Умеренная – от 15 до 20 % (900 -1200 ммол/л);
- Мягкая – от 10 до 15 мг % (600-900 ммол/л);
- Мягкая гиперфенилаланинемия – от 6 до 10 мг % (360-600 ммол/л);
- Мягкая гиперфенилаланинемия, при которой больные не нуждаются в лечении, — от 2 до 6 мг % (120-360 ммол/л).

Злокачественная гиперфенилаланинемия диагностируется при недостаточности тетрагидробиоптерина.
Лечение фенилкетонурии
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин. Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека.
Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются. Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ.
Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально!!!
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.

В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
В нашей стране на сегодняшний день единственным эффективным способом лечения является диетотерапия. Однако атипичные формы фенилкетонурии требуют постоянного приёма тетерагидробиоптерина или же его заменителей.
Питание
Чтобы нервные клетки малыша не подвергались токсическому действию фенилаланина и его производных, необходимо из рациона полностью исключить животные белки. Если сделать это на первых неделях жизни, то мозг ребёнка останется абсолютно здоровым. Если же ограничивать белок начинать в более позднем возрасте, то удаётся несколько приостановить задержку развития, однако устранить изменения в нервных клетках и вернуть здоровье нервной системе уже не удастся.
Соблюдать диету необходимо до 16 — 18 лет. Это является обязательным условием. Также желательно и в дальнейшем контролировать количество потребляемых животных белков.
Нужно придерживаться строгих правил касаемо грудного вскармливания. Необходимо использовать для кормления только сцеженное грудное молоко и лишь в количестве, разрешённом для определённого возраста. Для этого есть специальные таблицы, в которых приведены нормы потребления фенилаланина, а также существуют формулы расчёта количества молока в день. Кормящей матери при этом нужно также придерживаться специальной диеты.
Докармливают малыша специальными смесями, которые в своём составе не содержат фенилаланина. Введение прикорма у таких детей начинают с ягодных и фруктовых соков. В качестве твёрдой пищи ребёнку предлагают овощные пюре. Используют безбелковые крупы, а также безмолочные каши на основе кукурузной или рисовой муки.
Из меню детей дошкольного и школьного возраста исключают белковые продукты. Разрешены фрукты, овощи, растительные масла, изделия из крахмала.
| Возраст | Суточное количество фенилаланина (мг/кг) |
| менее 2 мес. | 60 |
| 2 — 3 мес. | 60 — 55 |
| 3 — 6 мес. | 55 — 45 |
| 6 — 12 мес. | 45 — 35 |
| 1 — 1,5 года | 35 — 30 |
| 1,5 — 3 года | 30 — 25 |
| 3 — 6 лет | 25 — 15 |
| старше 6 лет | 15 — 10 |

Также назначают витаминно-минеральные комплексы. Необходимо, чтобы ребёнок в нормальном количестве получал витамины B1, В6, С, фолиевую кислоту, кальций, железо и магний. Количество потребляемых калорий должно быть на 30% увеличено по сравнению с возрастной нормой.
Выделяют три группы натуральных продуктов, в основе которых лежит количество содержащегося в них фенилаланина.
- Красный список – это продукты, которые полностью необходимо исключить из рациона: все виды мяса и рыбы, морепродукты, колбасные изделия, яйца, творог, сыры, хлеб и хлебобулочные изделия, орехи, крупы и хлопья, кондитерские изделия, продукты из сои.
- Оранжевый список – продукты, которые под строгим контролем разрешены в небольших количествах: молочные продукты, овощи (картофель, капуста), овощные консервы, рис и кукуруза.
- Зелёный список – продукты, которые могут употребляться без каких-либо ограничений: овощи, фрукты, зелень, ягоды, крахмал, сахар, мёд, растительное и сливочное масло, топлёный жир.
Ещё две группы продуктов выпускает промышленность:
- готовые пюре на основе фруктов для детского питания;
- низкобелковые искусственные продукты для диетического питания (печенье, макароны, хлеб).
Родителям важно уметь правильно составлять диету и рассчитывать необходимое количество фенилаланина. Для этого нужно под рукой иметь весы, которые позволяют взвешивать до десятой доли грамма.
Своевременное лечение даёт хороший эффект. Ребёнка с ФКУ не госпитализируют. Родителям предоставляется квалифицированная консультативная помощь, малышу бесплатно выдают смеси, не содержащих животные протеины как часть специализированной диеты — единственного эффективного метода лечения ФКУ. Ребёнок получает специальные белковые гидролизаты с минимумом ФА или без него, но с достаточным уровнем витаминов и кальция.
При лечении грудное кормление ограничено (раньше полностью исключалось). Молоко сцеживают и дают согласно расчётам. Когда уровень фенилаланина нормализуется, ребёнка начинают прикармливать животным белком во избежание распада собственных тканей и истощения младенца. Включение фруктов, овощей, жиров, углеводов производится при строгом учёте уровня фенилаланина.
Сегодня не проблема найти диетический продукт или белковую смесь с определённой концентрацией ФА. По российскому законодательству дети, страдающие от ФКУ, получают бесплатное специальное питание. В РФ разрешены следующие составы:
- Афенилак.
- XP Аналог LCP.
- MD мил ФКУ-0.
Специальная диета строго соблюдается до 6 лет. Из рациона исключаются: хлеб, молочные продукты, яйца, рыба, мясо, а также Аспартам — искусственный подсластитель, который используют при изготовлении газированных напитков, конфет и жевательных резинок. Со временем нейроны становятся менее чувствительными к избытку ФА и после 16—18 лет подростки переходят на обычное питание. Но прекращение лечения может спровоцировать страх. Возвращение к диетотерапии улучшает психическое состояние пациентов.

У взрослых с фенилкетонурией наблюдаются лабильность настроения, депрессии. При высоких дозах ФА они страдают от деконцентрации и бессонницы, недостаточной мотивации, схематичности при принятии решений, импульсивности, потери способности рационально оценивать ситуации.
Лечение улучшает познавательные функции даже у страдающих тяжёлой умственной отсталостью. Если болезнь была поздно диагностирована, то соблюдение строгого режима питания позволяет снизить агрессивность, улучшить контакт пациентов с окружением. В некоторых случаях требуется пожизненное соблюдение диеты.
Лечение контролируют психиатр и медицинский генетик. При необходимости может понадобиться помощь логопеда. Больший эффект достигается при использовании ЛФК, массажа, иглорефлексотерапии.
Единственный способ лечения на сегодняшний день фенилкетонурии – диетотерапия, при соблюдении которой из питания исключаются продукты, содержащие аминокислоту фенилаланин.
Строгое соблюдение диеты однозначно необходимо, если уровень аминокислоты в сыворотке более 2-5 мг %, если этот показатель ниже, то лечебное питание не назначается.
Важное значение имеет срок назначения лечебного питания – нервная система малышей активно продолжает развиваться на протяжении первого года жизни и именно в это время необходимо уже начать терапию.

Соблюдение диеты позволяет вырастить полноценного ребенка без отклонений в умственном и психическом здоровье. Придерживаться эллиминационной диеты многие врачи советуют вплоть до 18-20 лет, то есть до момента окончательного формирования головного мозга.
После этого возраста питание можно расширять, но под контролем уровня аминокислоты в крови. При высоких уровнях фенилаланина (что устанавливается на основании анализов) диеты необходимо придерживаться до конца жизни.
Суть диеты – полный отказ от обычных белковых продуктов, больным детям нельзя давать:
- Мясо и содержащие его полуфабрикаты, рыбу;
- Морепродукты;
- Яйца;
- Творог и твердые сыры;
- Бобовые культуры;
- Муку из пшеницы (изделия из нее);
- Гречневую крупу, овсянку, манку.
Диета при фенилкетонурии должна отвечать следующим требованиям:
- Количество натурального белка в рацион должно быть сведено к минимуму;
- Белок (без фенилаланина) необходимый для нормального развития больной должен получать из специального лечебного питания;
- Организм обеспечивается энергией за счет использования низкобелковых продуктов;
- Соответствующую возрасту дозу микроэлементов и витаминов дети должны получать из дополнительных комплексов и лечебных средств для ФКУ.
У малышей около 80 % потребностей организма в белке должно восполняться за счет аминокислотных смесей, в составе которых нет фенилаланина.
До года это такие марки питания, как:
- Лофеналак;
- Нофемикс;
- Афенилак.
После года постепенно переходят на смеси:
- Нофелан;
- Фенилфри;
- Тетрафен;
- Бигрофен;
- Афенилак;
- МД мил ФКУ-3.
Прогноз
Проведение неонатального скрининга позволяет предотвратить серьёзные нарушения функции печени и повреждения нервной системы. При раннем назначении специальной диеты прогноз развития детей достаточно хороший. Однако стоит помнить, что при поздно начатом лечении в отношении умственного и психического развития ребёнка прогноз неблагоприятный.
Предупредить развитие необратимых повреждений органов ЦНС позволяет ранняя диетотерапия. Для того чтобы лечебное питание было назначено вовремя детям практически сразу после рождения проводится массовый скрининг и при необходимости назначается дополнительная диагностика.
Своевременное проведение элиминационной диеты и неукоснительное соблюдение предписаний врача по лечебному питанию позволяет родителям вырастить полностью здорового ребенка, то есть без отклонений в интеллектуальном, физическом и психическом развитии.
Прогноз течения патологии неблагоприятный при поздно начатой диетотерапии. Если в подростковом возрасте диета расширяется неадекватно или ее соблюдение полностью прекращается, то наблюдается снижение способности к обучаемости, нарушение поведенческих норм, расстройства со стороны психики.
Вот почему большинство психотерапевтов рекомендуют придерживаться элиминационной диеты как минимум до 18 лет.
Риск рождения детей, больных фенилкетонурией, оценивается после обследования супружеских пар в медико-генетических центрах. Обязательно такому обследованию должны подвергаться супруги, у которых уже рожден ребенок с таким заболеванием.
Желательно чтобы при планировании зачатия обследование на генетические патологии проходили пары, имеющие близкое кровное родство.
При атипичных формах ФКУ диетотерапия нужных результатов коррекции расщепления фенилаланина не дает. Прогноз течения таких типов патологии неутешительный – малыши либо гибнут в первые годы рождения, либо у них наблюдается глубокая умственная отсталость.
Фенилкетонурия пока единственное наследственное заболевание, раннее лечение которого предупреждает практически на 100 процентов вероятность развития тяжелых осложнений в будущем.
Поэтому родителям нельзя отказываться от неонатального скрининга их малыша в роддоме, а при рождении ребенка дома нужно его обследовать в течение первых трех недель жизни.
Очевидно, что данное заболевание требует немедленного принятия мер, в противном случае жизнь человека будет короткой.
Заболевание «фенилкетонурия» требует внимательного отношения к пациенту. Если соблюдать строгую диету и выполнять все рекомендации врачей, то ребенок сможет нормально расти и развиваться. Прогноз будет также зависеть от того, какие заболевания сопутствуют генетическому недугу и есть ли другие патологии.
Постепенно, с возрастом, организм может в какой-то мере приспосабливаться к повышенному содержанию фенилаланина, поэтому можно иногда допускать послабления в диете. Главное, чтобы не увлечься этими слабостями и вовремя остановиться и перейти на правильное питание.
Если женщина страдает этим недугом, то ей придется еще строже относиться к соблюдению всех рекомендаций, ведь она – будущая мама. Только в этом случае у нее есть возможность родить здорового ребенка.
Это особенно актуально, если учитывать, что методов профилактики данного заболевания практически не существует.
Устранить дефектный признак невозможно, поэтому своевременная диетотерапия помогает предотвратить развитие тяжёлых последствий. При планировании беременности генетический анализ назначается, если у родственников были случаи рождения детей с фенилкетонурией.
При своевременном выявлении и лечении у ФКУ благоприятный долгосрочный прогноз. Малыш в целом развивается нормально. Без диеты инвалидность наступает уже в первый год жизни. Соблюдение особого режима питания помогает предотвратить судорожные припадки и умственную неполноценность детей. Диета показана и будущим матерям с диагнозом «фенилкетонурия» во избежание микроцефалии, врождённых пороков сердца, слабоумия у ребёнка.
Сегодня разрабатываются и используются новые альтернативные методы лечения ФКУ, например, препаратом Сапроптерин для снижения уровня фенилаланина. Он эффективен при лёгких формах патологии. В России такие лекарства пока не применяются.
Народная медицина для избавления от недуга
Народные рецепты находят свое применение при лечении многих болезней. Не исключение и фенилкетонурия. Что это заболевание потребует пересмотра всего образа жизни – это факт. Ребенок должен расти и иметь представление о своем недуге. Родители обязаны в доступной форме объяснить ему, когда он будет способен осознавать полученную информацию, насколько серьезно его положение. Диету и лечение необходимо соблюдать в течение всей жизни. Только в этом случае можно гарантировать полноценное существование.
Народные лекари при фенилкетонурии рекомендуют употреблять больше растительных белков. В такой пище фенилаланина гораздо меньше, чем в животных продуктах. Не запрещается в рацион вводить фрукты и овощи. В них много витаминов и микроэлементов, которые в обязательном порядке необходимы для нормальной работы нервной системы. То есть народная медицина придерживается мнения, что такому больному желательно соблюдать вегетарианскую диету.
Питание при фенилкетонурии
Фенилаланин содержится практически во всех продуктах, в которых есть белок. Необходимо постараться исключить их из своего рациона, и в первую очередь это касается молока и мяса.
Если поставлен диагноз «фенилкетонурия», питание должно быть пересмотрено в первую очередь. Все продукты при этом можно разделить на несколько групп:
- Разрешены к использованию всегда: картофель, сахар, чай, растительные масла.
- Можно употреблять в небольшом количестве: рис, мед, масло сливочное, хлебобулочные изделия, овощи и фрукты.
Полностью необходимо исключить из своего меню: яйца, рыбу и мясо, молоко, макароны, бобовые, орехи, кукурузу, молочные продукты, шоколад.
Учитывая тот факт, что фенилаланин превращается в здоровом организме в тирозин, то больные фенилкетонурией должны в рацион включать продукты, содержащие его в достаточном количестве. К такой пище можно отнести грибы и растительные составляющие.
Исторические данные
Открытие данного заболевания связывают с именем Ивара Асбьёрна Фёллинга — норвежского врача, в 1934 году описавшего гиперфенилаланинемию, которую он ассоциировал с задержкой умственного развития. В честь открывателя патология также известна в Норвегии под названием «болезни Фёллинга».
Впервые успешное лечение было разработано и проведено в начале 50-х годов XX века группой медиков в Бирмингемском детском госпитале в Англии под руководством Хорста Биккеля. Но настоящий успех пришёл лишь после широкого применения так называемого метода Гатри, разработанного и внедрённого в 1958 — 1961 гг. (ранняя диагностика ФКУ по высокому содержанию фенилаланина в крови у новорождённых детей).
Со временем в диагностике и лечении данного заболевания стало ясно, что за него «отвечает» единственный ген — ген фенилаланингидроксилазы. Кроме того, описаны и выделены атипичные формы болезни, разработаны новые способы лечения, в ближайшей перспективе — генотерапия.
Заключение
Фенилкетонурия — опасная болезнь, но её можно победить. В этой борьбе многое зависит от родителей. Им приходится учиться мотивировать ребёнка к соблюдению диеты, а не злоупотреблять одними запретами. Если контролировать уровень ФА, избегать запрещённых продуктов, то особенный ребёнок сможет развиваться, реализуя всё то, что заложено в нём природой.
С целью оценки вероятности рождения малыша с фенилкетонурией должны пройти предварительное генетическое консультирование супружеские пары, уже имеющие такого ребёнка в семье, состоящие в родственном браке или имеющие родственников с фенилкетонурией. А женщины с этим заболеванием обязаны соблюдать строгую диету до и во время беременности.

По окончании университета прошла интернатуру по направлению «Неонатология» в СамГМУ. После завершения профессиональной подготовки, и по настоящее время, работаю врачом-неонатологом в ГУЗ Городская клиническая больница №1 (Перинатальный центр) г. Ульяновск.