
Оглавление
- 1 Что поражается в первую очередь
- 2 История заболевания
- 3 Как ставится диагноз
- 4 Историческая справка
- 5 Возможна ли профилактика болезни?
- 6 Почему проявляется генетический синдром?
- 7 Кто рискует больше всего
- 8 Классификация синдрома
- 9 Внешние проявления
- 10 Степень тяжести
- 11 Диагностика синдрома Марфана
- 12 Основное лечение
- 13 Осложнения на сердце
- 14 Прогноз и профилактика
- 15 Нарушение зрительной функции
Что поражается в первую очередь
Синдром Марфана — это нарушение нормального функционирования соединительной ткани. Поскольку она развивается неправильно, то это отражается на многих внутренних органах. У больных могут отмечаться следующие отклонения в организме:
- гигантизм;
- аневризмы аорты;
- долихостеномелия и арахнодактилия;
- миопия;
- эктопия хрусталика;
- кифосколиоз;
- деформация грудины;
- плоскостопие;
- эктазия твердой мозговой оболочки;
- протрузия вертлужной впадины.
Таким образом, есть общие физические особенности у людей, которым поставлен данный диагноз (синдром Марфана). Фото пациентов показывают сходство внешних признаков заболевания, и специалист уже по внешнему виду больного поймет достаточно много. Между тем при слабом проявлении этого заболевания внешние признаки могут быть практически незаметными.
История заболевания
К сожалению, на сегодняшний день вылечить синдром Марфана, как и повлиять на его причину, невозможно. Терапия в основном направлена на улучшение качества жизни больного человека, устранение симптомов и профилактику осложнений.
Лечение синдрома Марфана должно быть комплексным и может включать как консервативные, так и хирургические методики.
Всем пациентам с СМ рекомендуют ограничения в физической активности к среднему или низкому уровню, избегать тяжелого физического труда, не заниматься спортом, так как это способствует прогрессированию патологии и травматизму.
Больные должны наблюдаться у различных докторов: кардиолога, окулиста, травматолога-ортопеда, клинического генетика, невролога и др.

Лечением синдрома Марфана должна заниматься целая команда специалистов
В случае выявления тех или иных кардиоваскулярных проблем назначают комплексное лечение, медикаменты для профилактики осложнений. Проводят медикаментозную коррекцию аритмий, частоты сердечных сокращений, артериального давления. Обязательно всем пациентам назначают прием бета-блокаторов, если нет противопоказаний к этой группе средств, которые имеют протекторный эффект в отношении возможного расслоения аорты.
Рекомендуемые специалистами схемы терапии больных с СМ в обязательном порядке включают коллаген-образующие и метаболические средства, препараты для восполнения макро- и микроэлементов.
В случае проблем с органом зрения также проводят оперативные вмешательства при эктопии хрусталика, лазерную коррекцию зрения, подбирают очки или контактные линзы.
В целом подбор лечебных методик и спектр применяемых средств очень индивидуальны. Это полностью зависит от присутствующих симптомов и степени выраженности болезни у конкретного пациента.

Основная задача в лечении пациентов с Синдромом Марфана заключается в предотвращении сердечно-сосудистых повреждений
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие.
— гиперподвижность суставов;— аномалии строения тазобедренного сустава;— кифоз, сколиоз;— вывихи шейного сегмента позвоночника;— деформация грудной клетки;— плоскостопие;— глубокая посадка глаз;— уменьшенная нижняя челюсть, нарушение роста зубов;— высокое нёбо;— атрофические «растяжки» на коже;— паховые грыжи, частые разрывы связок.

— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);— пороки сердца (чаще — поражения клапанов);— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию.
Как мы уже отметили, для синдрома Марфана характерны:
- сочетанное поражение скелета, глаз, сердечно-сосудистой и нервной систем;
- многообразие физиологических проявлений;
- первые признаки заболевания могут быть диагностированы как при рождении ребенка, так и спустя годы;
- хроническое прогредиентное течение.
При этом заболевании наблюдаются стойкие нарушения функции суставов. Специалисты называют это состояние гипермобильностью, когда суставы легко выкручиваются в разные стороны и очень подвижны.
Многие пациенты, страдающие синдромом Марфана, имеют неправильное строение грудной клетки, она принимает воронкообразную или килевидную форму.
Ортопеды часто отмечают деформацию позвоночника в разных его проявлениях:
- сколиоз;
- кифосколиоз;
- кифоз;
- вывихи и подвывихи шейного отдела;
- спондилолистез.

Больные часто страдают плоскостопием и протрузией вертлужной впадины.
При синдроме Марфана высока вероятность внезапной смерти. Прогнозировать, сколько проживет человек с таким заболеванием, могут только специалисты, учитывая всю степень поражения организма. Своевременное лечение и кардиохирургическая коррекция позволяют улучшить качество жизни и увеличить ее продолжительность до 60-70 лет. При этом не стоит забывать о том, что люди с синдромом Марфана обязаны проходить диагностику и постоянно наблюдаться у доктора.
Медики ограничивают пациентам с этим заболеванием физическую активность, им противопоказаны высокие нагрузки, подводное плавание, занятия контактными видами спорта.
Женщинам, которые имеют в анамнезе это заболевание и планируют завести детей, необходимо пройти обследование у генетика.
Пациенты с синдромом Марфана имеют специфическую внешность, что упрощает диагностику заболевания. Обычно они имеют высокий рост, удлиненные конечности, при этом нижняя часть туловища длиннее верхней, а размах рук больше, чем рост. Астеничное телосложение сочетается с недостаточной массой тела, которая часто меньше 50 кг.
Людей с синдромом Марфана отличают характерные черты лица, что придает ему общее для всех пациентов данной группы «птичье» выражение. Глаза глубоко посажены, череп удлиненный, зубы скученны из-за малых размеров челюсти, готическое «небо». Нос кажется крупным, уши довольно большие и низко расположенные. У одного человека редко можно встретить все эти проявления, чаще всего среди внешних признаков отмечают только высокий рост и удлиненные пальцы.

Из-за слабости соединительных тканей у пациентов с синдромом Марфана могут наблюдаться стрии (растяжки на коже), которые не связаны с резкими скачками в весе или гормональными расстройствами. Эти симптомы проявляются обычно в области нижней трети спины, поясничного отдела и в зоне плеч.
Другой часто встречающийся симптом – грыжи, которые возникают самопроизвольно либо после операции, развиваясь из-за слабости соединительных тканей. Даже после хирургического лечения грыжа у больного с синдромом Марфана может появиться вновь.
-
Распространенный симптом со стороны органов зрения – подвывих хрусталика, который развивается из-за патологий соединительнотканных связок, а также миопия. У пациентов может наблюдаться отслоение сетчатки, вторичная глаукома, катаракта и другие глазные заболевания. Потеря зрения наряду с сердечнососудистыми патологиями является главной опасностью синдрома Марфана.
-
Со стороны опорно-двигательного аппарата часто наблюдаются искривления позвоночника по типу лордоза, кифоза и сколиоза, вдавливание и деформация грудной клетки, гиперподвижность суставов, гипотония мышц и плоскостопие.
-
Со стороны дыхательной системы часто появляются патологии, связанные с искривлением грудной клетки, в результате которого она сдавливает легкие. Повышен риск спонтанного пневмоторакса, что следует учитывать при занятиях спортом и физической активностью.
-
Со стороны сердечнососудистой системы наблюдают расслоение стенок аорты, патологии сердечных клапанов.
Что касается интеллектуального развития пациентов с синдромом Марфана, то оно не опускается ниже границ нормы, а в некоторых случаях может значительно превышать ее. Так, при среднем коэффициенте интеллекта в 85 единиц у больных с синдромом МарфанаIQ может быть больше 115, что является очень высоким показателем и встречается у одаренных людей.
Психические процессы при синдроме Марфана протекают своеобразно – дети с данным заболеванием могут быть гиперактивными, нервными и раздражительными, обладают завышенной самооценкой, болезненно реагируют на малейшие раздражители, часто плачут. Однако все эти проявления не указывают однозначно на наличие заболевания, а могут быть связаны с особенностями темперамента и протекания психических процессов.
Как ставится диагноз
Это заболевание в медицинской практике встречается нечасто. Как утверждают специалисты, на 10-20 тысяч человек только один может иметь синдром Марфана. Диагностика базируется на нескольких методах. Это семейный анамнез, а также исследования функций организма, офтальмологический осмотр, рентген и генетический прогноз.
Если доктор все же поставил диагноз “синдром Марфана”, лечение может быть как консервативным, так и хирургическим. К оперативному вмешательству приходится прибегать в серьезных случаях, когда лечение с помощью медикаментов и физиологических процедур не дает нужных результатов.
Оперативному вмешательству чаще всего подвергают сердечно-сосудистую систему и органы зрения.
Историческая справка

Первые упоминания о необычном недуге можно обнаружить в трудах американского офтальмолога Э. Вильямса, который в 1875 году описал признаки идентичного смещения хрусталиков глаз у родных брата и сестры. Кроме офтальмологических проблем эти дети имели повышенную подвижность суставов и высокий рост.
Известность болезнь приобрела позже, через 20 лет, когда французский педиатр Антуан Марфан представил свои наблюдения за 5-тилетей больной. Маленькая пациентка отличалась необычными аномалиями скелета и быстрым прогрессированием недуга. Синдром был назван в честь французского доктора, хотя впоследствии стало известно, что наблюдаемая им девочка страдала другой наследственной патологией – врождённой контрактурной арахнодактилией.
Можно найти множество примеров обнаружения синдрома у талантливых, знаменитых людей. Считается, что этим недугом страдали скрипач Никколо Паганини, американский президент Авраам Линкольн, русский композитор Сергей Рахманинов и другие известные личности. Некоторые исследователи считают, что неординарность людей с синдромом Марфана объясняется увеличенной концентрацией адреналина в крови. Этот гормон вызывает повышение активности и развитие незаурядных способностей.
Возможна ли профилактика болезни?
Генетическое заболевание очень редкое и недостаточно обследованное. Если говорить о причинах синдрома Марфана, самым правильным считается предположение о генетической мутации белка фибриллина. Это происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде. Можно выделить основные причины, способствующие появлению синдрома:
- наследственность;
- возраст отца (старше 35 лет).
Впервые болезнь детально описал в 1896 году французский педиатр А.Марфан, в честь которого и назвали патологию. Он наблюдал за 5-летней девочкой астенического телосложения с непропорционально длинными конечностями и врожденной арахнодактилией (длинными пальцами). К середине 20-х годов прошлого столетия уже имелось множество подобных описанных клинических случаев у детей и взрослых.
Непосредственная причина патологии в 95% случаев – это мутация в гене, которые кодирует строение фибриллина-1 и/или фибриллина-2. Локализируется мутация гена FBN1 и FBN2 в хромосоме 15 и 3.

Синдром Марфана наследуется по аутосомно-доминантному типу
Фибриллин – это основа эластических волокон соединительной ткани гликопротеиновой природы. Он составляет каркас межклеточного вещества, сосудистых стенок, хрящей, хрусталика глаза и многих других органов и тканей. В случае наличия описанной мутации у пациента соединительная ткань отличается повышенной способностью к растяжению, становится менее прочной и выносливой к механическим воздействиям, что и становится причиной клинических проявлений синдрома.
Примерно в 5% случаев непосредственной причиной синдрома Марфана (атипичные формы патологии) является точковая мутация гена, который кодирует строение α2-цепи коллагена первого типа.
К сожалению, специфической профилактики синдрома Марфана не существует. Семейная пара, в которой один или оба родителя страдают данной патологией, в обязательном порядке должны планировать беременность и пройти медико-генетическое консультирование у врача-генетика. Вместе с этим можно проводить пренатальную (дородовую) диагностику на предмет наличия СМ у плода.
Это аутосомно-доминантное состояние, генетическое заболевание, передается от родителей к ребенку через гены.
Вызван мутациями в гене FBN1. Мутации FBN1 связаны с широким континуумом физических функций, от изолированных особенностей до тяжелой и быстро прогрессирующей формы у новорожденных.
Особенности расстройства чаще всего обнаруживаются в сердце, кровеносных сосудах, костях, суставах и глазах. Некоторые особенности – например, расширение аорты (расширение основного кровеносного сосуда, которое переносит кровь от сердца к остальной части тела) – может быть опасным для жизни. Также могут быть затронуты легкие, кожа, нервная система. Патология не влияет на интеллект.
Распространенность, частота встречаемости составляет 1 из 5 000 человек, включая мужчин и женщин всех рас и этнических групп. Около 3 из 4 наследуют его, то есть получают генетическую мутацию от родителя, у которого она есть. Тип наследования при котором заболевший первый в семье- называется спонтанной мутацией. Существует 50-процентный шанс, рождения ребенка с генетической мутации от пораженных родителей.
Люди с синдромом Марфана рождаются вместе с ним, но особенности расстройства не всегда присутствуют сразу. У некоторых людей есть много особенностей при рождении, включая серьезные состояния, такие как расширение аорты. У других меньше симптомов, например, молодые не имеют признаков, пока не станут взрослыми. Некоторые особенности, особенно те, которые влияют на сердце и кровеносные сосуды, кости или суставы, со временем ухудшаются.
Это делает очень важным получение точной ранней диагностики и лечения. Без этого человек подвергается риску от потенциально опасных для жизни осложнений. Чем раньше началось лечение, тем лучше результаты.
Почти половина людей, страдающих синдромом Марфана, этого не знают.
Синдром Марфана – генетическое заболевание с наследуемостью аутосомно-доминантного типа, причиной его развития являются мутации гена FBN1. Этот ген отвечает за выработку фибриллина-1 – важного структурного белка, входящего в состав связок и эластичных сосудов. При нарушениях в строении этого белка соединительнотканные структуры становятся более растяжимыми, при этом теряют устойчивость к деформациям.
Наиболее опасные последствия таких изменений проявляются со стороны органов зрения и сердечнососудистой системы. Фибриллин-1 необходим для нормального функционирования цинновой связки, прикрепляющей хрусталик глаза к цилиарному телу. Поэтому при нарушении его синтеза, что происходит у больных с синдромом Марфана, цинновая связка ослабляется, что на первых этапах проявляется близорукостью у пациента, подвывихом хрусталика, но в дальнейшем может привести к вторичной глаукоме, частичной или полной потере зрения.
Другие функции фибриллина в организме – формирование внеклеточного матрикса, что обеспечивает целостность соединительных тканей и нормальное функционирование факторов роста клеток.
Аорта относится к сосудам эластического типа и содержит множество эластических связок, от которых зависит ее устойчивость к нагрузкам. При нарушении их функции возникает расширение аорты, расслоение ее стенок. Такая ситуация ставит под угрозу жизнь пациента, может спровоцировать внезапную смертность из-за разрыва аорты во время интенсивных физических нагрузок.
Среди других распространенных сердечных патологий, связанных с синдромом Марфана – поражения митрального клапана, из-за чего возникает необходимость хирургического лечения.
Синдром Марфана – генетическое заболевание, которое в 75% случаях наследуется по семейному типу, а оставшиеся 25% встречаемости являются результатом первичной мутации гена. В семьях, где отец старше 35 лет риск рождения ребенка с синдромом Марфана повышается.
Развитие симптомов начинается уже в период перинатального развития – соединительнотканные волокна в процессе формирования теряют прочность, с чего начинаются изменения в эластических сосудах, опорно-двигательном аппарате, глазных связках, твердом небе, сердечных клапанах. Прогрессирование заболевания продолжается в постнатальный период, и длится всю жизнь пациента.

Почему возникает синдром Марфана? Причины кроются в мутации гена FBN1. Именно он отвечает за синтез фибриллина. Этот структурный белок придает соединительной ткани нужную сократимость и эластичность. При дефиците фибриллина в организме плохо формируются волокнистые структуры. Соединительная ткань при этом теряет свою прочность и упругость. Она гораздо хуже выдерживает физиологические нагрузки. От этого начинают страдать внутренние органы и скелет человека.
Почему проявляется генетический синдром?
Причиной развития недуга считается мутация в гене FBN1, который располагается в 15 хромосоме и отвечает за нормальное производство фибриллина 1. Этот белков соединительной ткани является одним из главных компонентов, придающих ей эластичность и способность к сокращению.
Первыми при генетическом синдроме поражаются структуры, содержащие наибольшее количество важного белка – стенки кровеносных сосудов, связочный аппарат, цинновая связка глаза. Изменённая соединительная ткань не способна выполнять своей функции, выдерживать физическую нагрузку в связи с потерей прочности и упругости, у ребёнка возникают симптомы заболевания.
Недуг относится к генетическим и передаётся от родителей по аутосомно-доминантному типу. Риск появления малыша с наследственным синдромом очень высокий, если у мамы или папы имеются признаки болезни. В 75% случаях заболеваний прослеживается появление недуга в каждом поколении семьи. У 25% больных определяется новая, спонтанная мутация, не находится чёткой связи с наследованием.
Соединительная ткань не образует отдельного органа в человеческом теле. Но её клетки располагаются во всём организме. По средствам этих структур выполняются опорная, защитная и трофическая функции, образуется своеобразный каркас и покровы всех органов. К разновидностям соединительной ткани относят хрящевую, костную, мышечную, жировую ткани, кровь и лимфу. Поэтому системные заболевания, связанные с тканевой патологией, отличаются большим многообразием проявлений.
Кто рискует больше всего
Как уже отмечалось, синдром Марфана, признаки которого могут появиться уже в первые годы жизни человека, передается по наследству. Если в семье кто-то страдал этим генетическим заболеванием, высоки шансы, что ребенок может также унаследовать эту болезнь.
Но бывает и так, что у плода еще в утробе матери наблюдается первичная мутация. При этом чем старше роженица, тем больше риск рождения больного ребенка. Особенно это касается женщин, которые беременеют после 35 лет.
Классификация синдрома
Согласно МКБ-10, синдром Марфана входит в класс врожденных дефектов развития и хромосомных патологий, тут патологию можно найти под шифром Q87.4.
Детальной клинической классификации недуга на сегодняшний день не существует, но выделяют несколько форм болезни, в зависимости от тех или иных критериев.
В зависимости от выраженности симптомов:
- стертая форма – признаки патологии мало выражены и могут оставаться незамеченными на протяжении всей жизни, как правило, изменения касаются не более 2 систем органов;
- клинически выраженная форма – симптомы патологии хорошо заметны и встречаются более чем в 2 системах органов.

Внешний вид ребенка, больного синдромом Марфана
В зависимости от генетического фактора:
- семейная форма диагностируется в случаях, когда болезнь передается по наследству;
- спорадическая форма определяется тогда, когда патология обусловлена новой спонтанной мутацией у индивида и при этом не встречается у его родственников.
Болезнь отличается многообразием проявлением и различной их выраженностью. Этот недуг может быть длительное время нераспознанным, а некоторые отличительные особенности ребёнка расцениваться, как вариант нормы. В то же время существую формы, при которых характерные признаки болезни видны уже в роддоме.
Неонатальный вариант течения синдрома отличается выраженными признаками болезни у новорождённого, быстрым прогрессированием и высокой смертностью детей.
Признаки поражения органов незначительные и охватывают 1 – 2 системы организма;
Данную форму определяют, если хотя бы одна из систем организма имеет серьёзные нарушения функций. Врач может поставить этот диагноз и в случае обнаружения умеренных поражений 2 – 3 систем организма.
Большое значение в определении прогноза заболевание имеет динамика нарушений (специалисты различают прогрессирующий и стабильный варианты синдрома).
Формы патологии:
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
Этиологическая классификация:
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Внешние проявления
Чаще всего это заболевание заметно даже по внешним признакам. Причем чем старше становится пациент, тем более явно проявляются внешние отличительные черты. У людей, которые имеют в анамнезе синдром Марфана, фото, как уже говорилось, очень схожи.
Их скелет имеет определенные особенности — туловище относительно короткое, рост высокий, конечности длинные и тонкие, непропорциональные скелету. Пальцы также удлиненные, паукообразные.
Синдром марфана признаки имеет и такие:
- астеническое телосложение;
- слаборазвитая подкожная клетчатка;
- мышечная гипотония;
- узкий и длинный лицевой скелет (долихоцефалия);
- высокое аркообразное небо, а также нарушение прикуса (прогнатии).
При рождении ребенка с синдромом Марфана длина тела у мальчиков составляет свыше 53 см, у девочек — 52,5 см. Окончательный рост соответственно наблюдается в районе 192 и 175 см. Бывают и более высокие показатели.
Степень тяжести
Это заболевание имеет разные степени поражения. Соответственно, у людей, которым диагностирован синдром Марфана, симптомы могут быть разные.
Медики выделяют несколько разных форм этого заболевания в зависимости от того, какие жизненные системы поражены.
Первая форма — стертая. Изменения и отклонения в развитии слабо выражены. Страдают только одна иди две системы организма, причем не очень сильно.
Вторая форма синдрома Мафрана – выраженная. Здесь имеется несколько вариантов:
- Изменения выражены слабо, но присутствуют в трех системах организма.
- В одной системе наблюдаются явные отклонения от нормы.
- Изменения четко выражены в двух, трех и более системах.
Медики отмечают также три степени тяжести заболевания: легкую, среднюю и тяжелую. Течение болезни также может быть разным. Есть стабильный синдром Марфана, признаки которого на протяжении многолетних наблюдений остаются неизменными. Есть прогрессирующий тип заболевания, когда с течением времени патологии в развитии нарастают и усугубляются.
Диагностика синдрома Марфана
Синдром Марфана диагностируется на основании данных физикального осмотра, семейного анамнеза, обследования у кардиолога и офтальмолога, рентгенологических исследований органов грудной клетки.
Немаловажную роль при диагностике играют такие критерии:
-
Наличие дефектов грудной клетки, которые требуют хирургического вмешательства;
-
Наличие патологических изменений тканей и органов зрительной, сердечнососудистой системы, характерных для синдрома Марфана;
-
Эктопия хрусталика;
-
Верхняя часть туловища значительно короче нижней, их соотношение больше 0,86;
-
Размах рук больше, чем рост ( их соотношение больше 1,05);
При синдроме Марфана в несколько раз возрастает выведение глюкозаминов через почки, что также используется при диагностике.
ЭКГ и эхокардиография позволяют обнаружить нарушения сердечной деятельности. Для обнаружения патологий со стороны дыхательной системы проводят обследование внешнего дыхания. У пациентов с синдромом Марфана часто наблюдается вздутие легочных тканей, гиперкапния, неравномерное распределение воздуха по легким и разнообразные нарушения механики дыхания.
Исследования ДНК (прямое автоматическое секвентирование) позволяют определить наличие мутаций в гене FBN1, идентифицировать их.
Диагностика при синдроме Марфана носит в основном клинический характер. Обязательно учитывают анамнез, в том числе и семейный (наличие подобных проблем у кого-то из родственников), данные объективного обследования и осмотра. Также проводят множество дополнительных диагностических процедур для выявления патологии тех или иных органов и систем.
Для этого применяют ЭКГ, УЗИ сердца и сосудов, рентгенографию органов грудной клетки, КТ, МРТ внутренних органов, позвоночника, головного мозга, офтальмоскопию и прочие исследования органа зрения, аортографию, ангиографию и много других методик, в зависимости от клинической ситуации и симптомов болезни.
Существуют общепринятые диагностические критерии синдрома Марфана (большие и малые), которые позволяют с большой степенью вероятности поставить правильный, но предварительный диагноз пациенту.
Окончательный диагноз синдрома Марфана выставляют только после анализа генотипа (ДНК-диагностика) и выявления специфической мутации в гене, ответственном за продукцию фибриллина, с помощью молекулярно-генетических методик.
Выявлением синдрома Марфана занимаются специалисты в области генетики, кардиологии, офтальмологии, неврологии, ортопедии. Диагностика патологии включает сбор анамнеза жизни и болезни, выявление типичных клинических признаков, анализ внешнего осмотра и физикальных данных, результатов кардиографического и рентгенографического обследования, посещения офтальмолога, составление родословной у генетика.
Основные диагностические методики:
- общий анализ крови и мочи — типичные признаки воспаления;
- биохимическое исследование крови позволяет выявить дисфункцию определенного органа, возникшую в результате развития патологического процесса, а также определить первопричину заболевания и назначить правильное лечение;
- электрокардиография отображает электрические потенциалы, сформированные в работающем сердце;
- эхокардиография – исследование морфологических и функциональных изменений сердца и его клапанного аппарата;
- рентгенографическое и томографическое исследование — информативные диагностические методики, обнаруживающие поражение костей, суставов, внутренних органов и мягких тканей;
- аортография – рентгенологического исследования аорты с использованием контрастного вещества;
- УЗИ внутренних органов,
- биомикроскопия и офтальмоскопия,
- молекулярно-генетический анализ.
Для правильной постановки диагноза малышу придётся пройти комплексное обследование, получить консультацию у многих специалистов.
Поскольку патология имеет наследственную природу, в большинстве случаев удаётся проследить заболевание в семье. Нужно помнить, что недуг может протекать в различных формах и многие люди, с лёгким течение синдрома, не догадываются о своём заболевании на протяжении всей жизни.
Поскольку болезней, связанных с патологией соединительной ткани, существует множество, иногда бывает сложно правильно поставить диагноз ребёнку. Чтобы справится с этой задачей в 1996 году генетиками и клиницистами были разработаны современные критерии, с помощью которых можно определить синдром Марфана.
К ним относятся:
- увеличение роста в большей степени за счёт верхней части тела;
- грубая деформация грудной клетки и позвоночника;
- продольное плоскостопие;
- невозможность полностью разогнуть конечность в коленных и локтевых суставах (контрактуры);
- эктопия хрусталика;
- расширение и расслоение восходящей части аорты и другие признаки.
Некоторые из этих симптомов можно встретить у абсолютно здоровых детей. В диагностике генетического синдрома большую роль играет именно их сочетание. Триада Марфана включает в себя патологию костно-суставной системы, органические изменения в сердце или крупных сосудах, болезни глаз;
Эти признаки в меньшей степени указывают на наследственный дефект, но их наличие и сочетание с большими критериями подтверждает диагноз синдром Марфана.
К ним относятся:
- высокая подвижность суставов;
- аномалии зубов, нёба;
- гипоплазия радужной оболочки глаза, цилиарной мышцы, увеличение длины глазного яблока;
- пролапс митрального клапана;
- патологии бронхо-лёгочной системы, спонтанный пневмоторакс и другие нарушения.
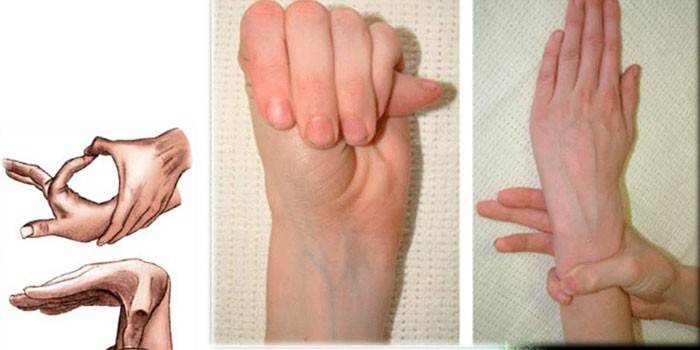
Врач просит ребёнка обхватить запястье одной руки большим пальцем и мизинцем другой руки, образуя «браслет». В пользу наследственного недуга говорит лёгкое смыкание кисти на запястье другой руки, нахождение фаланг мизинца и большого пальца друг на друга;
Исследователь просит малыша попытаться дотянутся большим пальцем до предплечья этой же руки. Тест считается положительным, если ногтевая фаланга пальца ребёнок с лёгкостью достаёт до лучевой кости предплечья.
Обычные клинические и биохимические анализы крови и мочи не показательны при синдроме Марфана, изменений в них может не быть. Помочь в установлении диагноза поможет обнаружение продуктов метаболизма соединительной ткани в моче.
Резкое увеличение оксипролина и гликозаминогликанов в суточной моче может говорить о развитии осложнений у ребёнка (прогрессировании сердечной недостаточности, отслойке плаценты, развитии пневмоторакса, пневмонии).
С помощью современных методов исследования, молекулярно-генетической диагностики можно обнаружить характерную для данного генетического синдрома мутацию в гене FBN1.
Такое заболевание, как синдром Билса отличается от синдрома Марфана дефектом синтеза другого белка – фибриллина 2, но клинические симптомы этих недугов схожи. Различить их можно только с помощью молеклярно-генетической диагностики и выявления особого симптома «мятого уха», что указывает на наличие синдрома Билса.
Специфические проявления заболевания можно обнаружить во многих органах, поэтому заподозрив у ребёнка наследственный синдром, проводятся различные исследования. Патологию костно-суставной системы обнаруживают с помощью рентгенографии и компьютерной томографии.
Болезни сердца и сосудов диагностируются благодаря ЭКГ и ЭхоКС, МРТ. С помощью УЗИ обнаруживаются патологии внутренних органов, смещение их положения в брюшной полости. В исследовании органа зрения помогут такие методы, как офтальмоскопия, биомикроскопия.
Ребёнок с наследственным синдромом состоит на учёте у многих врачей: генетика, травматолога, хирурга, офтальмолога, кардиолога и других специалистов.
Диагностика синдрома Марфана у детей младшего и среднего возраста затруднена. Это объясняется быстрым ростом ребёнка и изменением нарушений. Выраженность некоторых патологий при правильном лечении уменьшается, в то же время при отсутствии необходимой терапии клинические проявления становятся более яркими, появляются новые симптомы. Поэтому стоит сохранять настороженность в отношении детей с подозрением на генетический синдром, регулярно обследовать и лечить малыша.
Для определения точного диагноза синдрома паучьих пальцев врачи собирают семейный анамнез, проводят внешний осмотр, обязательно назначают генетическое исследование, КГ, ЭхоКГ и так далее. Специалисты определяют соотношение роста человека и его кистей, выявляют деформацию грудной клетки, кифосколиоз, наличие долихостеномелии, проводят тесты Варги, охвата запястья и так далее.
Благодаря рентгенографии врачи обнаруживают увеличенные размеры левого желудочка, диагностируют протрузию вертлужной впадины, расширение дуги аорты. Для диагностики синдрома Марфана часто используют ЭхоКГ, на котором видны пролапс митрального клапана, дилатация аорты. Если есть подозрение на проблемы с аортой, назначается аортография, для диагностирования эктопии хрусталика – офтальмоскопию. При диагностике синдрома паучьих пальцев специалисты сравнивают болезнь с другими.
Основное лечение
Арахнодактилия не лечится. Симптомы, появляющиеся по мере прогрессирования долихостеномелии, устраняются разными способами. Главная цель медикаментозной терапии – не допустить развития осложнений. Если, к примеру, на первом году жизни у ребенка диагностировали марфаноподобный фенотип, аневризму аорты, сразу назначают средства для предотвращения ее прогрессирования.
Лечение синдрома Марфана заключается и в коррекции зрения. Больному подбирают правильный очки, в сложных случаях используют лазерный или хирургический методы. Если у ребенка marfan syndrome, наблюдаются скелетные нарушения, рекомендованы торакопластика, эндопротезирование. Курс лечения включает метаболическую терапию, прием витаминов, препаратов с глюкозамина сульфатами, янтарной кислоты, при слишком быстром росте – гормонов. Людям с марфаноидным фенотипом стертой формы показаны физические занятия.
Лечение синдрома Марфана имеет симптоматический характер и направлено на уменьшение негативных проявлений заболевания со стороны сердечнососудистой, зрительной и других систем.
С целью профилактики осложнений больным рекомендуется избегать видов спорта, связанных с резкими бросками и рывками, как баскетбол, бега на спринтерские дистанции, контактных видов спорта, во время которых возможны телесные повреждения. Тем не менее, не следует отказываться от спорта вовсе, в группу риска попадают такие занятия, которые вызывают резкое изменение сердечного ритма, частоты сердечных сокращений. Если контролировать свой пульс во время занятий спортом и не выходить за рамки безопасных показателей, то такая активность не противопоказана.
С раннего возраста рекомендуется принимать лекарственные препараты, снижающие нагрузку на аорту, как бета-адреноблокаторы. В составе комплексного лечения детям с синдромом Марфана прописывают витаминные комплексы, антиоксиданты и энерготропные средства курсами продолжительностью в месяц по несколько раз в году.
К ним относятся:
-
Токоферол, аскорбиновая кислота, витамины группы B;
-
Рибоксин в таблетках;
-
КоэнзимQ10;
-
Элькар;
-
Лимонтар курсом в десять дней из расчета 5 мг на 1 кг веса;
-
Димефосфон.
Из ноотропных препаратов назначают пирацетам по 400 мг дважды в сутки курсом в два месяца, повторяют 3-4 раза в год.
Хирургическое вмешательство при кардиальных симптомах заболевания требуется в случае, если корень аорты у пациента достигает 5 см и больше.
В течение всей жизни больной с синдромом Марфана должен постоянно проходить обследования у специалистов различного профиля – офтальмологов, кардиологов, ортопедов и генетиков, так как заболевание проявляет себя по-разному в каждом отдельном случае, провоцируя нарушения функции различных органов и систем.
Кроме того, необходимо санировать очаги инфекции в ротовой полости с применением антибиотиков, так как у больных с синдромом Марфана ослабленный иммунитет. Антибиотикотерапию в целях профилактики используют даже при незначительных хирургических вмешательствах, чтобы снизить вероятность развития инфекционного эндокардита.
Хирургической коррекции подлежат не только патологии сердца, расслоение стенок аорты и поражения клапанов, но и заболевания внутренних органов и скелета, возникающие в ходе прогрессирования синдрома Марфана.
Бета-адреноблокаторы применяются не только в целях снижения нагрузки на сердечнососудистую систему, но и эффективно убирают негативные проявления артериальной гипертензии и аритмии, характерные для данного заболевания.
‹
Интерфероны: плацебо или лекарство? Мифы и научные факты!
9 мифов о низкоуглеводных диетах
›
Специфической терапии, направленной на устранение причины заболевания не существует. В настоящее время не разработаны методы влияния на наследственный аппарат клетки. Поэтому основная цель лечения синдрома Марфана – предотвращение прогрессирования заболевания, борьба с симптомами болезни:
- патология сердца и сосудов.
Самым опасным проявлением недуга считается аневризма, расширение участка аорты. Коварство болезни заключается в непрерывном, длительном прогрессировании симптомов. Случается, что опасный симптом формируется к 18 годам, поэтому важно уделять достаточно внимания ежегодному обследованию и лечению сердечно-сосудистой системы.
Грубые пороки развития сердца и сосудов, тяжёлые осложнения хронических болезней лечатся оперативно. Из лекарственных препаратов назначаются ингибиторы АПФ, блокаторы кальциевых каналов. Применение b-адреноблокаторов (пропанолола, атенолола) показано при расширении корня аорты, пролапсе клапанов, аритмиях.
Назначение препаратов и подбор необходимой дозировки должно проводиться врачом-кардиологом с учётом данных обследования ребёнка. Необоснованное назначение лекарственных средств может привести к ухудшению состояния ребёнка;
- болезни опорно-двигательного аппарата.
Существую исследовании, указывающие на дефицит некоторых макроэлементов (кальций, цинк, кобальт, магний) и белков, необходимых для строительства соединительной ткани при синдроме Марфана. Поэтому для предотвращения прогрессирования патологии назначаются витаминно-минеральные комплексы, гиалуроновая кислота, викасол, колекальциферол. Оперативное лечение показано при грубых патологиях развития скелета;
- заболевания органов зрения.
Исправление патологии зрения проводится с помощью подбора специальных очков, контактных линз, оперативного лечения катаракты и глаукомы, смещения хрусталика.
Опасным осложнением синдрома является отслойка сетчатки. Эта патологии возникает при активном занятии спортом у ребят с дефектом соединительной ткани. Повышенная физическая нагрузка, прыжки, травмы приводят к отделению тонкой сетчатой оболочки от сосудистой. Такое нарушение сопровождается резким снижением остроты зрения, которое не всегда является обратимым. Поэтому ребятам с генетическим синдромом следует избегать чересчур активных занятий, для таких детей хорошо подходит плавание в бассейне;
- нарушенный обмен веществ.
Для улучшения метаболизма рекомендовано использовать в комплексном лечении аскорбиновую и янтарную кислоты, карнитин, препараты магния, токоферола ацетата. С целью нормализации обмена хрящевой ткани используются глюкозаминсульфат, хондроитинсульфат.
Существуют данные, указывающие на необходимость введения диеты с высоким содержанием магния для детей с синдромом Марфана. Этот элемент помогает справиться с повышенным содержанием катехоламинов в крови и способствует восстановлению дефектов кровеносных сосудов. Хорошо включать в ежедневный рацион орехи, какао, гречневую и ячневую каши, сухофрукты.
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.
Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.

Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Стоит отметить, что при этом заболевании лечение не может назначить один врач. Ведут наблюдение и вырабатывают рекомендации специалисты разного профиля: офтальмолог, ортопед, кардиолог, кардиохирург, терапевт, генетик.
В первую очередь лечение направлено на профилактику прогрессирования этого заболевания. Поскольку избавиться от болезни окончательно невозможно, важно остановить ее, чтобы в дальнейшем не поражались жизненно важные органы.
Если имеется недостаточность клапанов сердца, то показана хирургическая операция. При острой необходимости ставится протез митрального клапана.
Беременные женщины, у которых синдром Марфана имеет явно выраженную сердечно-сосудистую патологию, направляются на досрочное кесарево сечение.
Если нарушено зрение, то специалисты подбирает специальные очки и контактные линзы. При явных патологиях, когда поставлены такие диагнозы, как катаракта, глаукома, смещение хрусталика, назначается хирургическое или лазерное лечение.

Также операция показана, если явно выражены нарушения в скелетной системе. Доктора проводят стабилизацию позвоночника, торакопластику, эндопротезирование тазобедренных суставов.
Осложнения на сердце
В клинической картине этого заболевания часто доминирует сердечно-сосудистая патология. Она проявляется по-разному. У пациента могут наблюдаться стойкие дефекты стенок кровеносных сосудов. Они теряют свою эластичность. Особенно этому подвержены аорты, крупные ветви легочной артерии. У больных могут наблюдаться пороки развития клапанного аппарата и перегородок сердца.
Что касается аорты, то здесь чаще всего наблюдаются прогрессирующие расширения ее восходящей части и клапанного кольца (дилатация, аннулоаортальная эктазия). У больного часто имеются аневризмы, поражен митральный клапан. Наблюдается также патологическое удлинение створочных хорд и их разрыв.
У ребенка, который еще находится в утробе матери, но на генном уровне синдром Марфана ему уже передался, часто формируются врожденные пороки сердца. Это могут быть:
- коарктация аорт;
- дефект межпредсердной перегородки (ДМПП);
- стеноз легочной артерии;
- дефект межжелудочковой перегородки (ДМЖП).
Также может наблюдаться нарушение сердечного ритма (тахикардия, фибрилляция предсердий), развитие инфекционного эндокардита.
При самом неблагоприятном варианте неонатальной формы синдрома Марфана сердечная недостаточность быстро прогрессирует с самого рождения ребенка. Часто такие дети не доживают до одного года.
Прогноз и профилактика
Синдром Марфана отличается, как правило, хроническим прогрессирующим течением. Продолжительность жизни пациентов при условии полноценного комплекса лечебных мероприятий в среднем составляет 45 лет. Основными факторами риска преждевременной смерти выступают осложнения, которые возникают вследствие патологии кардиоваскулярной системы.
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.

Течение болезни во многом зависит от выраженности клинических проявлений и качества проведённого лечения. Особую опасность для жизни ребёнка представляют пороки развития сердца и сосудов, их патологические изменения, поэтому терапии этой группы заболеваний отводится особое место.
Родителям девочек нужно знать об опасности будущей беременности для здоровья больной наследственным синдромом. Именно во время вынашивания ребёнка увеличивается риск развития и расслаивания аневризмы аорты. Это связано с повышенной нагрузкой на систему кровообращения и гормональными изменениями.
В целом, при правильном лечении и вовремя оказанной помощи пациенты с генетическим синдромом доживают до глубокой старости. Болезнь привносит ограничения в выборе будущей профессии. Таким детям лучше искать занятие не связанное с повышенной физической нагрузкой, поднятием тяжестей.
Профилактика недуга заключается в своевременном диагностировании синдрома в семье и медико-генетическом консультировании будущих родителей.
Нарушение зрительной функции
Пациенты с этим заболеванием в большинстве случаев страдают патологией органа зрения. Это могут быть:
- близорукость;
- уплощение и увеличение размера роговицы;
- вывих/подвывих хрусталика;
- гипоплазия радужной оболочки и цилиарной мышцы;
- изменение калибра сосудов сетчатки;
- косоглазие.

При этом в возрасте до 4 лет часто развивается эктопия хрусталика, она быстро прогрессирует, зрительная функция ухудшается.